Saúde
Terapia genética vence doença sanguínea hereditária
Pacientes de talassemia têm menos hemoglobina no sangue que o normal, resultando em anemia crônica, com consequências até fatais. Novo método – já aprovado na Europa – elimina necessidade de transfusões frequentes
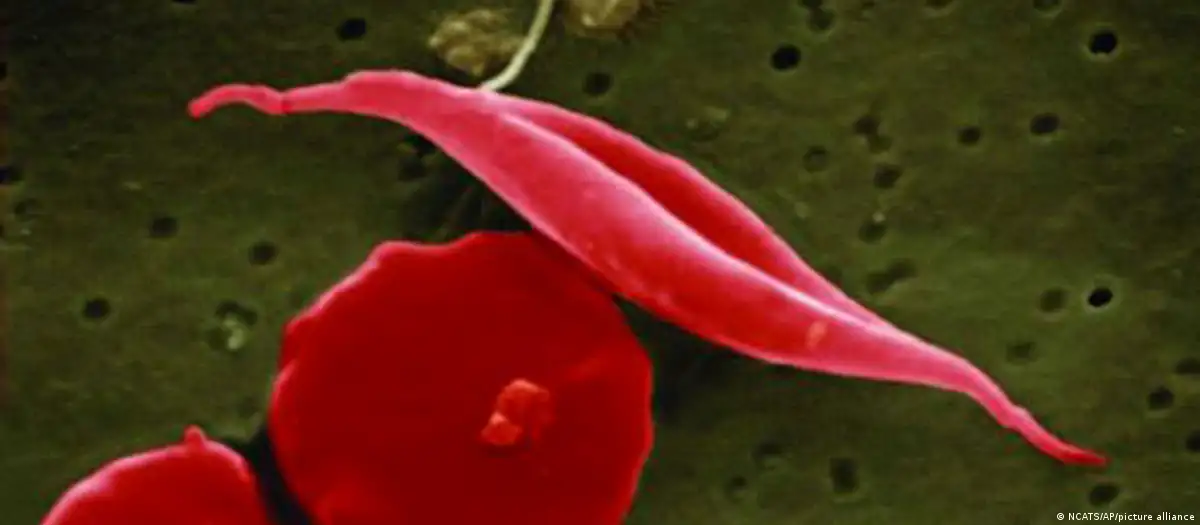
A talassemia e a anemia falciforme (também denominada drepanocitose ou anemia drepanocítica) são distúrbios hereditários que resultam numa redução da hemoglobina nos glóbulos vermelhos do sangue. São doenças bastante difundidas, atingindo cerca de 7% da população mundial, e desencadeadas pela mutação de um único gene.
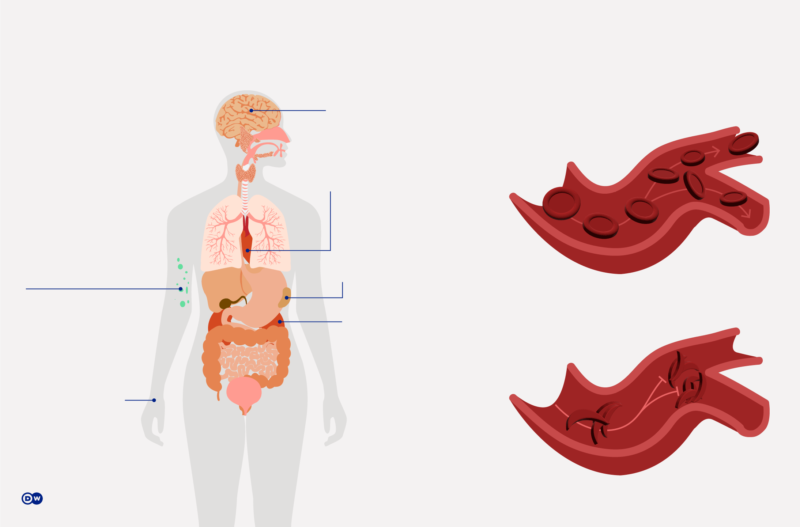
No organismo saudável, as hemácias se movem sem problemas através dos menores vasos sanguíneos. O composto proteico hemoglobina, que lhes dá a cor vermelha, contém ferro e é responsável pelo transporte do oxigênio.
Na talassemia e na anemia falciforme, um defeito genético reduz a capacidade e o tempo de vida dos glóbulos vermelhos. Nos pacientes de anemia falciforme, eles assumem forma de foice ou de lua crescente, perdem a elasticidade e obstruem os vasos sanguíneos, que se inflamam.
Em ambos os casos, o resultado é anemia crônica, causando dores e danos aos órgãos e tecidos privados do oxigênio necessário. Entre os possíveis sintomas resultantes, estão inchaço pronunciado do fígado e do baço, alteração dos espaços medulares e deformações do esqueleto. Sem terapia adequada, a anemia pode ser fatal para as crianças de até cinco anos de idade.
Revolução que vem das células-tronco
Até o momento, os únicos tratamentos possíveis para a talassemia e a anemia falciforme eram transfusões de sangue regulares, por toda a vida, ou transplante de células-tronco. No entanto é muito difícil encontrar material de transplante apropriado para cada um dos pacientes que, apesar de ambas as terapias, continuam tendo uma expectativa de vida drasticamente reduzida.
A terapia para talassemia com a tesoura genética CRISPR/Cas9, desenvolvida por 15 clínicas da Europa e dos Estados Unidos, promete agora vida basicamente normal e saudável para os portadores de talassemia. Ela foi testada em pacientes entre 12 e 35 anos de idade, dos quais mais de 90% já vivem há mais de 12 meses sem necessidade de transfusões, tendo sido publicada no New England Journal of Medicine.
O tratamento pode durar de alguns meses a um ano: são retiradas do sangue células-tronco, e modificadas com a tesoura CRISPR/Cas9, de modo a produzirem hemoglobina fetal sem defeitos e plenamente funcional.
Depois que a medula do paciente é “apagada” através de uma quimioterapia, são reimplantadas as células modificadas, que passam a produzir hemácias saudáveis, eliminando a necessidade de transfusões. A Agência Europeia de Medicamentos (EMA) já liberou para doentes a partir de 12 anos essa primeira terapia genética bem-sucedida.

Ao longo do “cinturão da malária”
A talassemia se manifesta sobretudo na Índia, Paquistão, Bangladesh, Afeganistão, China Meridional, Sudeste Asiático, Península Árabe, Iraque, África Ocidental e Setentrional, na Turquia, região do Mar Negro e Mediterrâneo.
Curioso é que a distribuição da talassemia no mundo corresponde, grosso modo, à do “cinturão da malária” histórico. Os pacientes da enfermidade genética, de fato, resistem melhor ao parasita plasmódio, causador da malária, por possuírem mais glóbulos vermelhos, mesmo se com menos hemoglobina.
O novo método genético não substituirá inteiramente o transplante de células-tronco, explica Peter Lang, diretor da Clínica de Medicina Infanto-Juvenil de Tübingen, Alemanha: “A terapia é, antes, para os enfermos para quem não se encontrou doador ou que, por algum motivo, não podem receber células-tronco alheias. Pode ser também realizada naqueles que já se submeteram a um transplante e depois tiveram uma recaída.”
Assim como a de Düsseldorf, a Clínica da Universidade de Tübingen está entre as 15 que participaram da pesquisa. Para Lang, “nossa terapia é um exemplo fantástico de que tratamentos genéticos são eficazes e podem ser empregados no dia a dia clínico”.
")