Saúde
Anvisa se reúne domingo para definir autorização emergencial de vacina
Confira o status das análises preliminares para registro e outras informações de vacinas
A Diretoria Colegiada da Anvisa – Agência Nacional de Vigilância Sanitária deve se reunir no próximo domingo, 17, para discutir os pedidos de autorização para uso emergencial de vacinas contra a covid-19 e bater o martelo sobre as solicitações. Segundo comunicado oficial da agência, a data é o penúltimo dos 10 dias estipulados como limite para este tipo de exame pela agência reguladora.
O Instituto Butantan, em parceria com a farmacêutica chinesa Sinovac, e a Fiocruz, parceira do consórcio Astrazeneca/Oxford, entraram com requerimentos de autorização em caráter emergencial para suas respectivas vacinas.
Em seu site, a Anvisa passou a divulgar o status das análises preliminares para registro e outras informações de vacinas contra covid-19. Clique aqui para abrir em alta resolução.
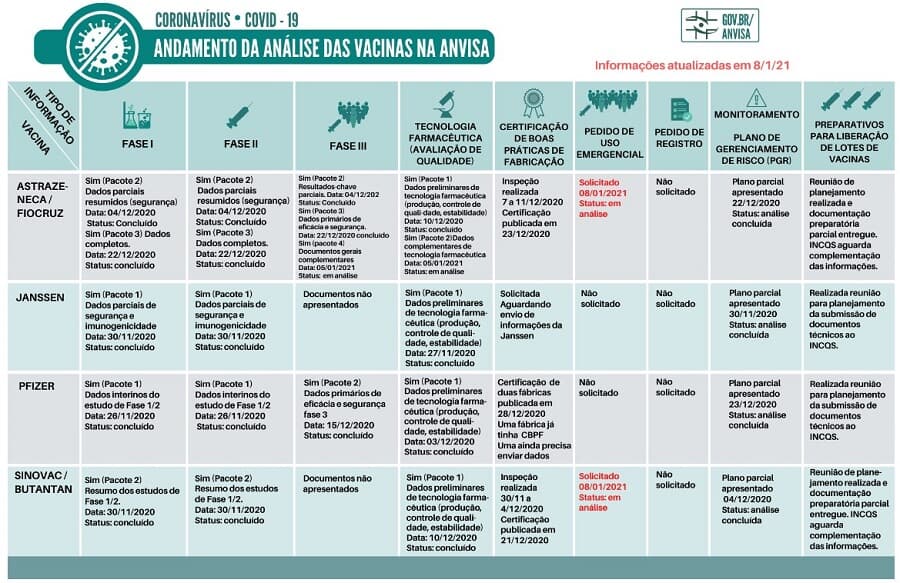
A vacina Sputnik V não aparece no quadro de análise da Anvisa porque o pedido de anuência do estudo ainda está em avaliação. A agência ainda não a considera como uma vacina em teste no Brasil.
Entenda cada uma das fases
Fase 1 – essa é a primeira etapa de testes em humanos para avaliar a segurança e possíveis reações indesejáveis no local da aplicação da vacina ou no organismo. Nessa fase também pode ser verificada, de forma preliminar, a imunogenicidade da vacina, ou seja, sua capacidade de gerar anticorpos contra o novo coronavírus.
Fase 2 – é hora de avaliar a dose, a forma de vacinação e a capacidade de gerar anticorpos (contra o novo coronavírus) na população (faixa etária, por exemplo) que deverá ser indicada para receber a vacina. A segurança continua em análise aqui.
Fase 3 – os testes nessa etapa são realizados em grandes populações para avaliar a segurança e a eficácia da vacina. A vacina precisa provar que, de fato, é capaz de nos proteger da doença.
Pedido de uso emergencial – feito antes do registro final para aplicar a vacina em um grupo específico da população. Precisa ser enviado à Anvisa pela empresa fabricante ou importadora da vacina, presente no território brasileiro. Pode ser realizado com a fase 3 em andamento.
Registro – profissionais especializados da Agência vão revisar todos os documentos técnicos e regulatórios e verificar os dados de segurança e eficácia, bem como avaliar a qualidade da vacina. O registro concedido pela Anvisa é o sinal verde para que a vacina seja comercializada e disponibilizada no país. Precisa ser solicitado à Anvisa pela empresa fabricante da vacina.
Dados de eficácia e segurança – essas informações compreendem estudos clínicos e não clínicos. Estão incluídos aqui dados referentes aos estudos realizados em animais e humanos.
Dados de tecnologia farmacêutica – são dados referentes à qualidade, ao processo de fabricação e ao controle de qualidade. Eles têm como objetivo demonstrar que o produto será fabricado com qualidade e de forma consistente, dentro das especificações de uso. Esses dados também determinam o prazo de validade do produto.
Pacotes – são grupos de documentos apresentados pelas empresas à Anvisa.
Pacotes 1, 2, 3 e sucessivamente – são pacotes com dados e informações parciais. As empresas apresentarão mais dados e informações em pacotes subsequentes ou quando apresentarem o pedido de registro da respectiva vacina.
Status – é referente à análise de um determinado pacote de documentos.
Submissão parcial – assim chamada porque a empresa não apresentou a documentação completa para o registro da respectiva vacina.
Certificação de Boas Práticas de Fabricação – avaliação realizada por inspetores qualificados para garantir que uma fábrica, em qualquer lugar do mundo, cumpre com os requisitos determinados pela legislação brasileira. São avaliadas as estruturas físicas das áreas de produção, armazenamento e laboratórios de controle de qualidade, além de toda a documentação do sistema de garantia de qualidade da empresa.
Dados
Conforme a última atualização, do início da noite desta terça-feira, 12, 33,7% da documentação entregue pelo Butantan estava pendente de complementação; 5,4% ainda não haviam sido apresentados; 40,17% foram concluídos e 20,13% estão em análise.
Entre os pontos que precisam de material adicional está o desfecho da análise de eficácia do estudo clínico da fase 3 (a última etapa) e testes de pureza, identidade e potência da vacina, além de dados de segurança e eficácia em subgrupos de pacientes por status de infecção. Hoje, o Instituto divulgou informações adicionais sobre a eficácia da vacina.
Já no caso da vacina da Astrazeneca/Oxford/Fiocruz, 14,4% do material repassado à Anvisa precisa ser complementado; 32,39% foram concluídos e 53,17% estão em análise. Entre os pontos que necessitam de mais informações está o processo de fabricação, incluindo atributos críticos de qualidade.
")